
Abstract
PURPOSE Patients are frequently asked to share their personal health information. The objective of this study was to compare the effects on patient experiences of 3 electronic consent (e-consent) versions asking patients to share their health records for research.
METHODS A multi-arm randomized controlled trial was conducted from November 2017 through November 2018. Adult patients (n = 734) were recruited from 4 family medicine clinics in Florida. Using a tablet computer, participants were randomized to (1) a standard e-consent (standard), (2) an e-consent containing standard information plus hyperlinks to additional interactive details (interactive), or (3) an e-consent containing standard information, interactive hyperlinks, and factual messages about data protections and researcher training (trust-enhanced). Satisfaction (1 to 5), subjective understanding (0 to 100), and other outcomes were measured immediately, at 1 week, and at 6 months.
RESULTS A majority of participants (94%) consented to future uses of their health record information for research. No differences in study outcomes between versions were observed at immediate or 1-week follow-up. At 6-month follow-up, compared with the standard e-consent, participants who used the interactive e-consent reported greater satisfaction (B = 0.43; SE = 0.09; P <.001) and subjective understanding (B = 18.04; SE = 2.58; P <.001). At 6-month follow-up, compared with the interactive e-consent, participants who used the trust-enhanced e-consent reported greater satisfaction (B = 0.9; SE = 1.0; P <.001) and subjective understanding (B = 32.2; SE = 2.6, P <.001).
CONCLUSIONS Patients who used e-consents with interactive research details and trust-enhancing messages reported higher satisfaction and understanding at 6-month follow-up. Research institutions should consider developing and further validating e-consents that interactively deliver information beyond that required by federal regulations, including facts that may enhance patient trust in research.
INTRODUCTION
Growing use of electronic health record (EHR) data for research has created practical and ethical challenges for health systems and researchers.1,2-4 In 2018, the Federal Policy for the Protection of Human Subjects (Common Rule) was updated to permit health care institutions to seek broad consent for unspecified, future research5 involving secondary uses of protected health information. However, the relatively vague nature of broad consent for future, yet-to-be-determined studies presents an implementation challenge in ensuring that broad consent is informed consent.
Informed consent involves patients making voluntary decisions based on accurate and desired information about research procedures, risks, protections, and potential benefits. Moreover, patient trust—a key component of all interactions between patients and health care institutions6,7—is critical for patient decisions about participating in research.8-10 Trust facilitates patient comprehension of and engagement with information during the consent process.11 In current practice, though, informed consent processes may not always include facts that patients prefer to receive or that help patients accurately assess the trustworthiness of research processes.12 For example, facts about research regulations, researcher training processes, and data protections may not be presented to patients as they consider the likelihood and potential negative impact of research risks.13 In addition, previous research has not rigorously examined the long-term effect of providing such information in informed consent processes.
The objective of our study was to compare the effects of 3 electronic consent (e-consent) versions with varying degrees of interactive information and factual messages intended to enhance patient trust.
METHODS
We conducted a multi-arm, parallel, single-blinded, randomized controlled trial to compare 3 tablet-based e-consent interventions, each asking patients to consider agreeing to include their EHR data in a family medicine database for use in future, yet-to-be-specified research studies. Reporting of this study follows the Consolidated Standards of Reporting Trials (CONSORT) 2010 Statement and the CONSORT extension statement on reporting of multi-arm parallel-group trials.14,15 The University of Florida’s institutional review board approved this study.
Setting and Participants
Participants were adult patients making regularly scheduled visits to 1 of 4 family medicine clinic locations affiliated with the University of Florida academic health center. The e-consent intervention and immediate follow-up survey were administered onsite at participating clinics. We conducted additional follow-up by phone at 1 week and 6 months post-consent. Eligible patients were aged 18 years or older and able to communicate in English.
Procedures
We recruited participants from November 2017 through March 2018. Participants received $25 USD for completing the e-consent intervention and immediate follow-up assessment, and $15 for each additional follow-up survey.
Study staff approached patients in clinic waiting rooms. Potential participants were asked to review a brief information sheet explaining that the research team was building a database of patients willing to share their health records for researchers to use in future studies. We disclosed to participants that, although the e-consent was being tested as part of a research project, their decision to share their health records for future research was real, not hypothetical.
Once enrolled, participants were given a tablet computer and randomized to 1 of the 3 e-consent versions. Participants were instructed to complete the e-consent independently with a study team member on hand to assist with technical difficulties. Immediately after participants completed the e-consent, they were prompted on the tablet to complete a follow-up survey to assess outcomes and demographic characteristics. All study outcomes were assessed again at 1 week and 6 months with follow-up by oral telephone survey. The 1-week follow-up survey could be completed up to 2 weeks and the 6-month follow-up survey up to 7 months after the participant’s enrollment. We made 3 contact attempts before considering a participant lost to follow-up. Immediate follow-up surveys were administered from November 2017 through March 2018; 1-week follow-up surveys from November 2017 through April 2018; and 6-month follow-up surveys from May 2018 through November 2018.
Interventions
The e-consent versions were developed and piloted through a user-centered design process intended to identify and incorporate content and design elements important to patients.13 Each e-consent version was tablet-based, self-guided, written at an 8th-grade reading level, and concluded by asking participants for consent to include their EHR data in a family medicine research database for future research studies.
Version 1: Standard e-Consent
The standard e-consent contained all federally required elements of informed consent,5 including descriptions of the purposes of the research; procedures to be followed; any reasonably foreseeable risks or discomforts to the participant; any benefits to the participant or others which may reasonably be expected from the research; the extent to which confidentiality of records identifying the participant will be maintained; and a statement that participation is voluntary (Supplemental Figure 1, https://www.AnnFamMed.org/content/19/1/16/suppl/DC1/). We refer to this version as the standard e-consent because it contained the standard, minimum level of information required for obtaining informed consent as mandated by federal law.
Version 2: Interactive e-Consent
In clinical settings, the informed consent process typically consists of a face-to-face interaction to explain the purpose and potential risks and benefits of the research procedure, during which potential participants may ask questions as desired. However, in the case of a broad consenting process intended to administer consent to all patients at a systems level, face-to-face interaction is impractical. Therefore, we designed an interactive e-consent containing hyperlinks that each patient could click to discover additional information on demand without being overwhelmed by a larger volume of less personally relevant information. The interactive e-consent was identical in content and length to the standard e-consent except for the addition of interactive hyperlinks on key terms. Using these hyperlinks, participants could access additional information, including research study examples, explanations of EHR content, and terminology definitions (Supplemental Figure 2, https://www.AnnFamMed.org/content/19/1/16/suppl/DC1/).
Version 3: Trust-Enhanced e-Consent
The trust-enhanced e-consent contained all elements present in the standard and interactive e-consents with the addition of factual messages designed to explain trust-relevant attributes of research institutions and processes. Specifically, the messages conveyed information on research regulations, researcher training processes, and data protection procedures (Supplemental Figure 3, https://www.AnnFamMed.org/content/19/1/16/suppl/DC1/). Because of this additional information, the trust-enhanced e-consent was slightly longer than the interactive and standard versions.
Randomization
We randomly allocated participants to 1 of the 3 study e-consent versions in a parallel 1:1:1 scheme. Because the e-consents did not support automated randomization, study staff members were not blinded to participant assignment. The randomization sequence was generated by a statistician and occurred at the individual participant level.
Outcomes and Measures
The primary outcomes were satisfaction with the consent decision and subjective understanding of consent information. We assessed satisfaction using the Satisfaction with Decision scale, a validated instrument measuring patient satisfaction with a health care decision.16 Participants were asked to rate their satisfaction (5-point scale; “strongly disagree” to “strongly agree”) with various aspects of their decision to consent or not for future research use of their health record data (eg, “I am satisfied that I am adequately informed about the issues important to my decision”). Subjective understanding was measured using the Subjective Understanding subscale of the Quality of Informed Consent (QuIC) scale.17 Participants were asked to rate their understanding of key general aspects of research participation on a scale from 1 (“I didn’t understand this at all”) to 5 (“I understood this very well”), with final scores ranging from 0 to 100. Because the QuIC was developed in the context of clinical trials, we modified the wording where necessary to reflect the focus of our study; for example, we changed the statement “the treatments and procedures you will undergo” to “what will happen to your health record data.”
Secondary outcomes were objective knowledge of study details, perceived voluntariness, and trust in medical researchers. We measured objective knowledge (scale: 0 to 6) by asking participants to respond “agree,” “disagree,” or “unsure” to 6 statements developed by the research team that reflected factual information present in all 3 e-consent versions (eg, “I know that this study was about letting researchers study my personal health information, such as my blood pressure numbers or medications I may have taken”). Perceived voluntariness (scale: 9 to 45) was measured using the Decision-Making Control Instrument, a validated tool designed to measure the extent to which participants view treatment and research decisions in clinical settings as voluntary.18 Finally, trust was assessed using the Trust in Medical Researchers Scale, a validated instrument with summary scores ranging from 0 to 48.19 We asked participants to rate their agreement (5-point scale; “strongly disagree” to “strongly agree”) with statements related to medical researcher intentions (eg, “To get people to take part in a study, medical researchers usually do not explain all of the dangers about participation”).
In addition, we collected participant sociodemographic characteristics, including age, sex, race, ethnicity, and highest level of education. We also recorded participants’ decision to share their health record data (agree/disagree).
Sample Size Calculation
Sample size calculations were completed a priori for the primary outcome measures. The target sample size of 600 participants with 10% expected attrition at 1 week was designed to have over 85% power to detect a difference in averages across immediate and 1 week primary outcomes between versions of 1.1 units for satisfaction with consent decision and 2.8 units for subjective understanding of consent, under 2-sided hypothesis testing and an α of 0.05. During the trial, additional resources became available to increase our recruitment target and therefore statistical power. Ultimately, we enrolled 750 participants.
Statistical Analysis
We computed descriptive statistics for participant characteristics and primary and secondary outcomes for the overall sample and by e-consent version and follow-up time point. Next, linear mixed models were used to assess the impact of e-consent version, time point, a variable interacting e-consent version with time point, and participant age, education level, sex, race, and ethnicity. The model for each outcome included a participant random effect and fixed effects for e-consent version, follow-up time point, and each demographic covariate.
Sensitivity analyses were conducted to investigate the possibility that participants lost to 6-month follow-up differed in important ways from participants remaining in the final analytic sample. An inverse probability of dropout weighted analysis was conducted for all primary and secondary outcomes, in which 1-week and immediate outcomes, baseline covariates, and e-consent version allocation were used to predict the probability of remaining in the study at 6-month follow-up. The reciprocal of the probability of remaining in the study were then applied as weights to data from 6-month follow-up only; results of linear regression analyses from these weighted data were found to be similar to non-weighted results, indicating little evidence for important differences between dropouts and remaining participants.
For all analyses, we used SAS, Version 9.4 (SAS Institute Inc) statistical software. Statistical significance was assessed at the α = 0.05 level.
RESULTS
A total of 1,242 patients were approached, 750 (60.4%) agreed to participate, and 734 completed the e-consent and immediate follow-up assessment (Figure 1). We observed no statistically significant differences in participant characteristics between e-consent versions (Table 1). In addition, participants were demographically similar to the underlying clinic population except for the proportion of Black patients (43.3% in this study vs 30.6% in the clinic population).

CONSORT flow diagram of participant randomization, allocation, follow-up, and analysis.
CONSORT = Consolidated Standards of Reporting Trials
Participant Characteristics by e-Consent Version
Approximately 94% (n = 691) of participants agreed within the e-consent to share their EHR data with the family medicine database for use in future research, with no statistically significant differences in consent decision observed between e-consent versions (94.6% standard, 93.6% interactive; 94.6% trust-enhanced).
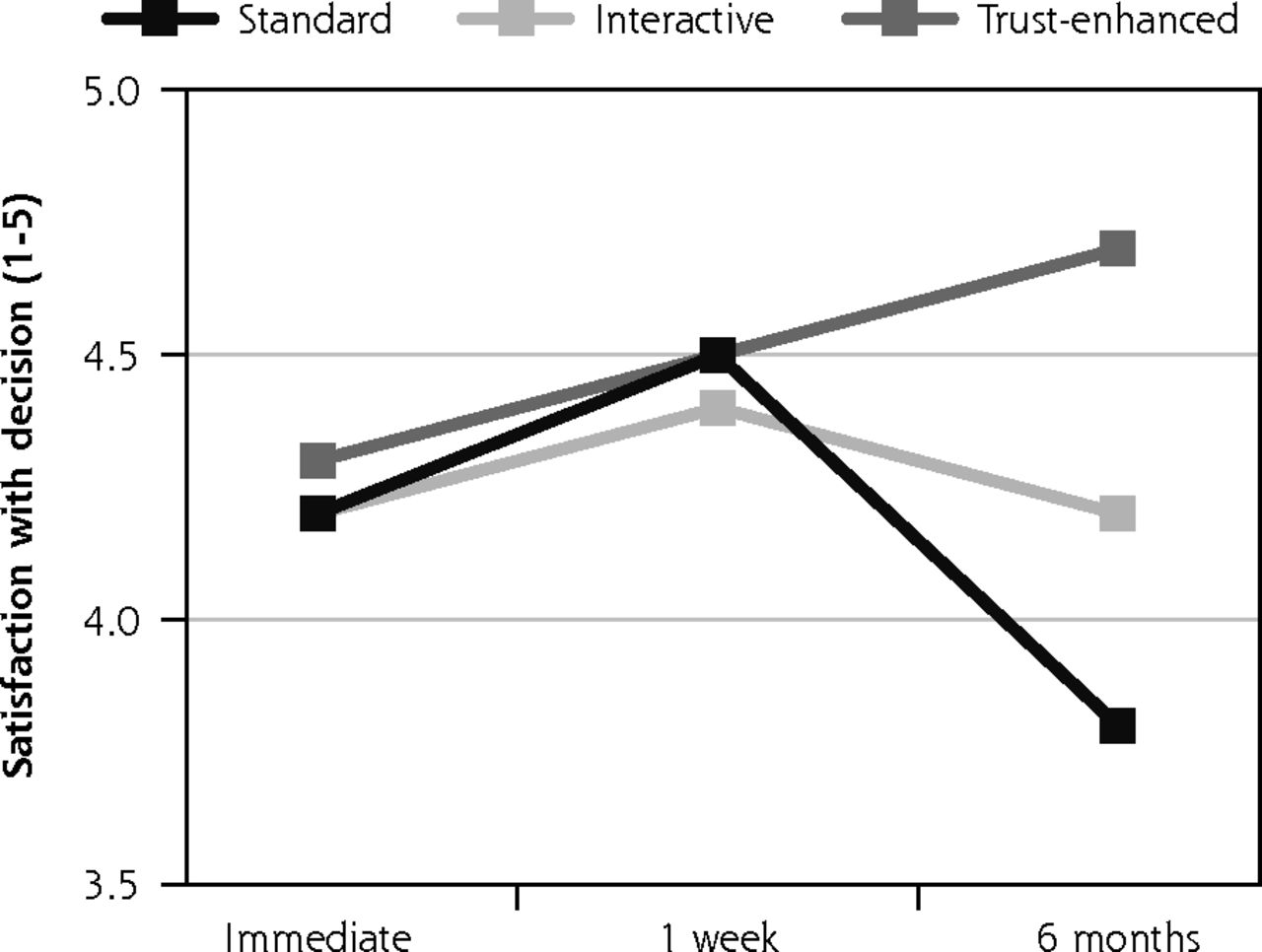
Descriptive analyses of outcomes by e-consent version revealed that mean satisfaction with the consent decision was similar across the 3 versions at immediate follow-up and 1-week follow-up. However, at 6-month follow-up, mean satisfaction decreased to 3.8 (SD = 0.7) for the standard e-consent, remained similar for the interactive version at 4.2 (0.4) and increased for the trust-enhanced version to 4.7 (0.5) (Figure 2).

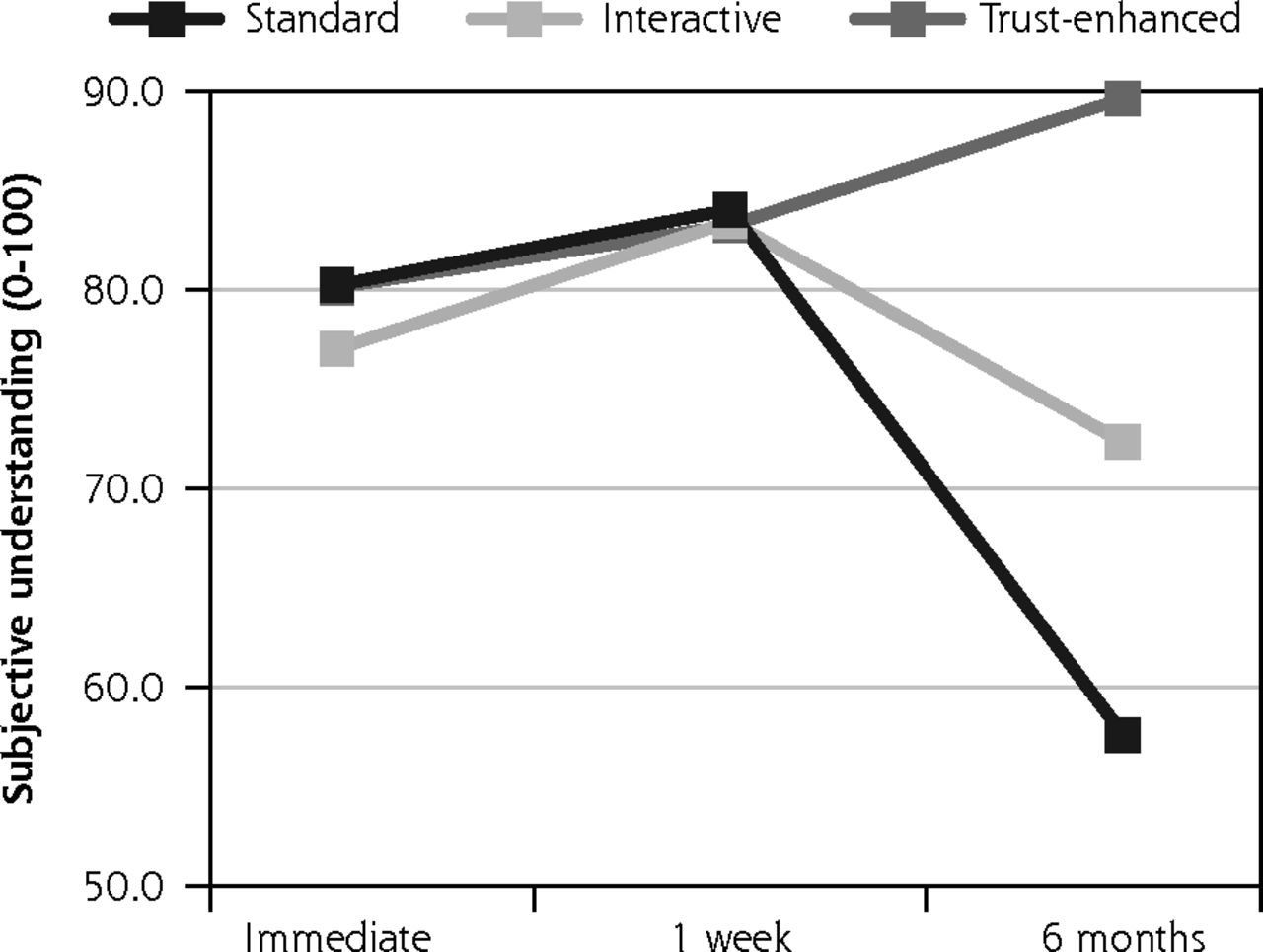
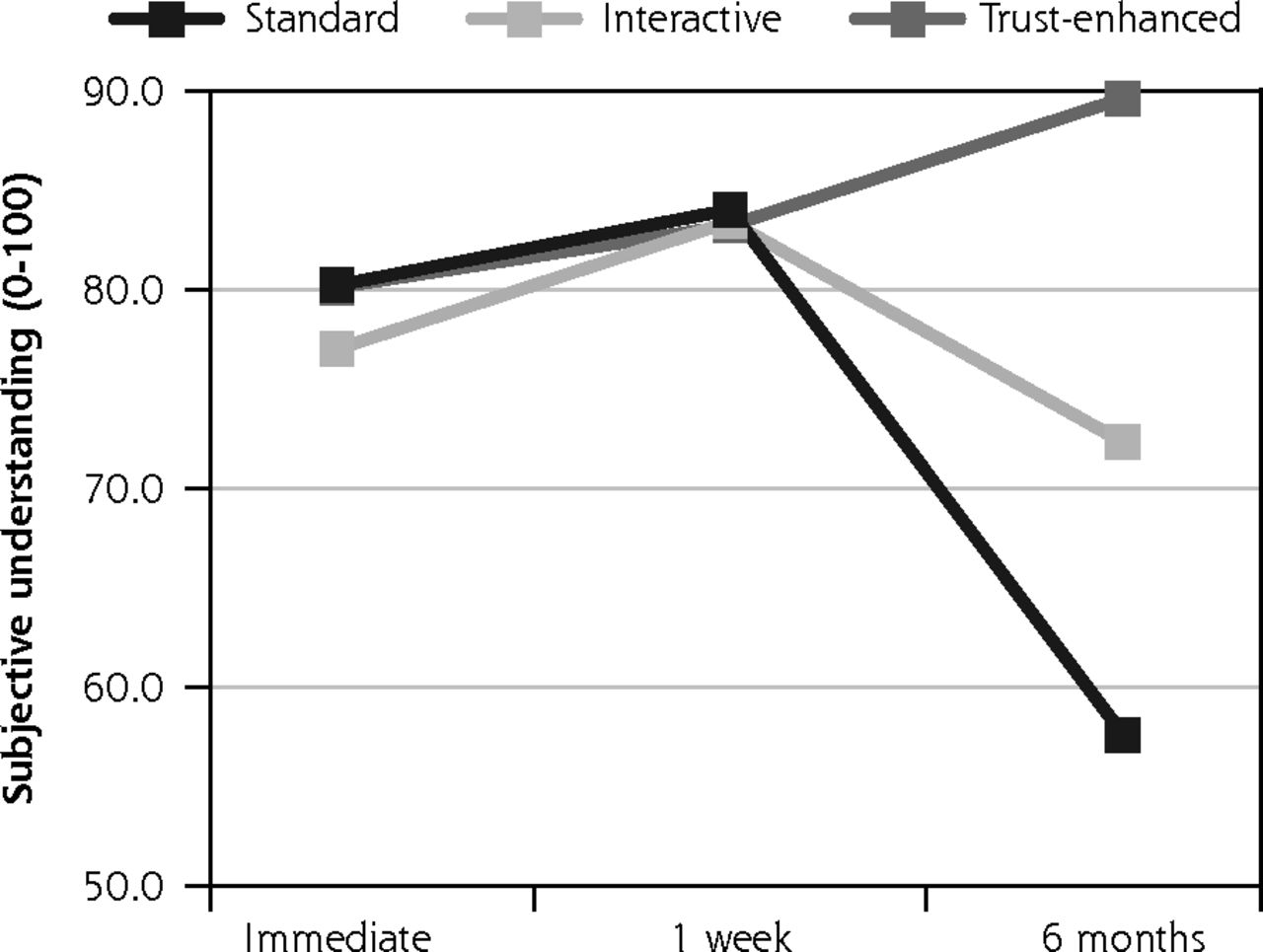
Primary outcomes: satisfaction with decision and subjective understanding.
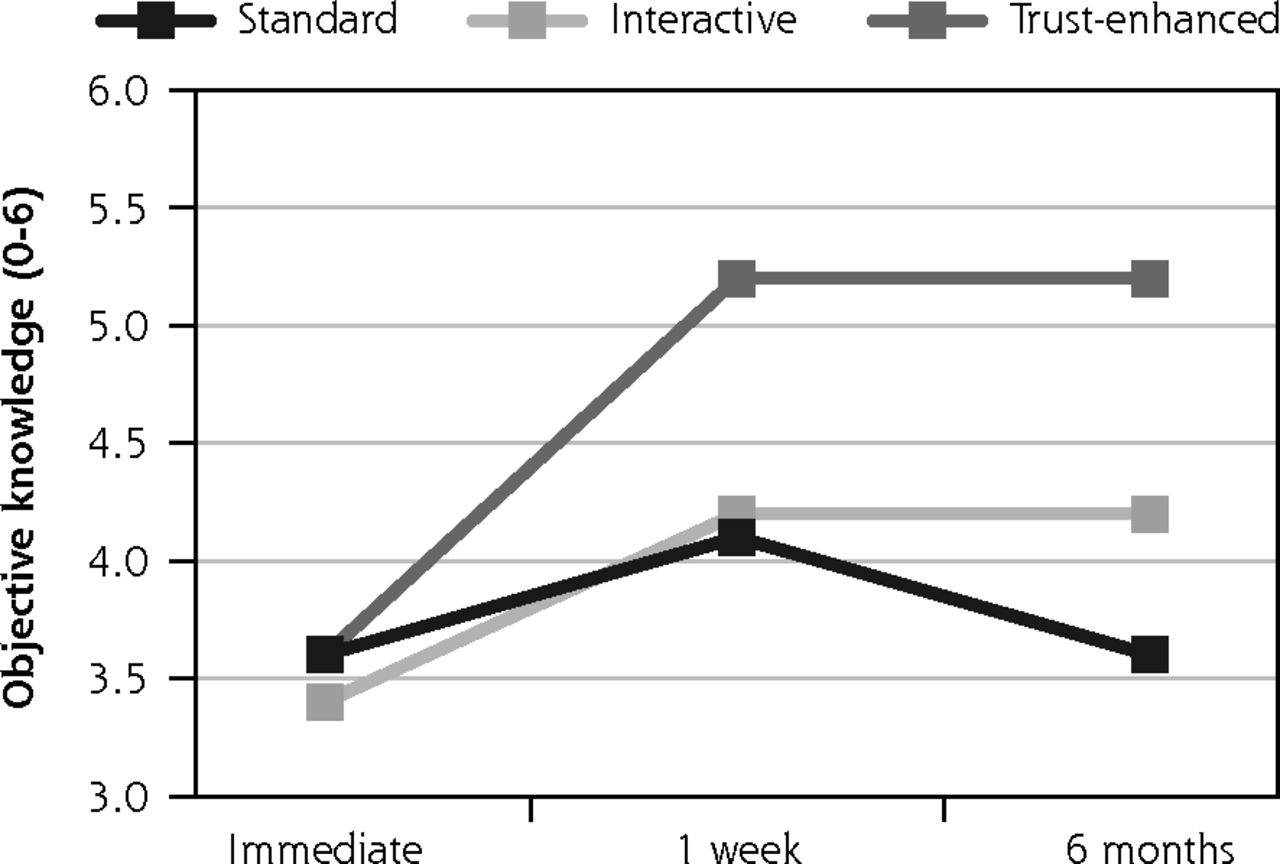
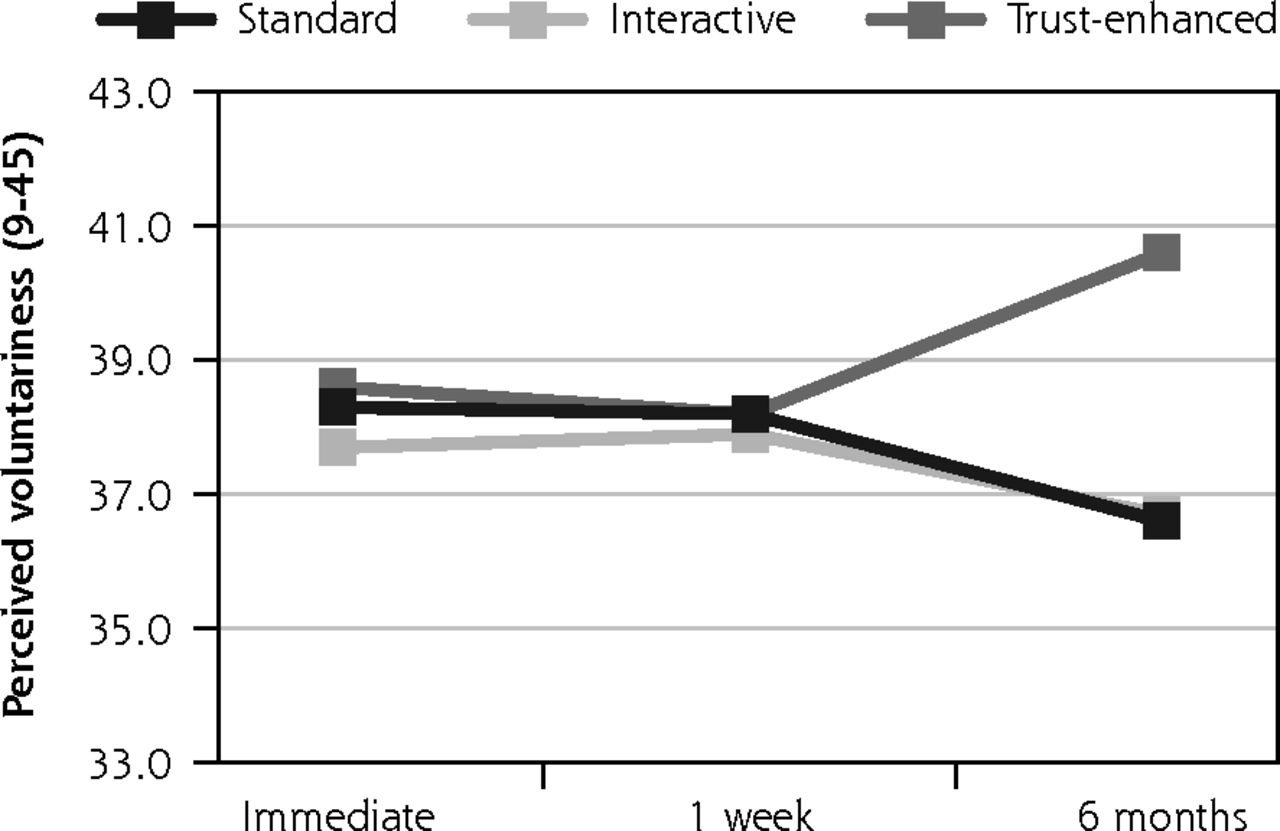

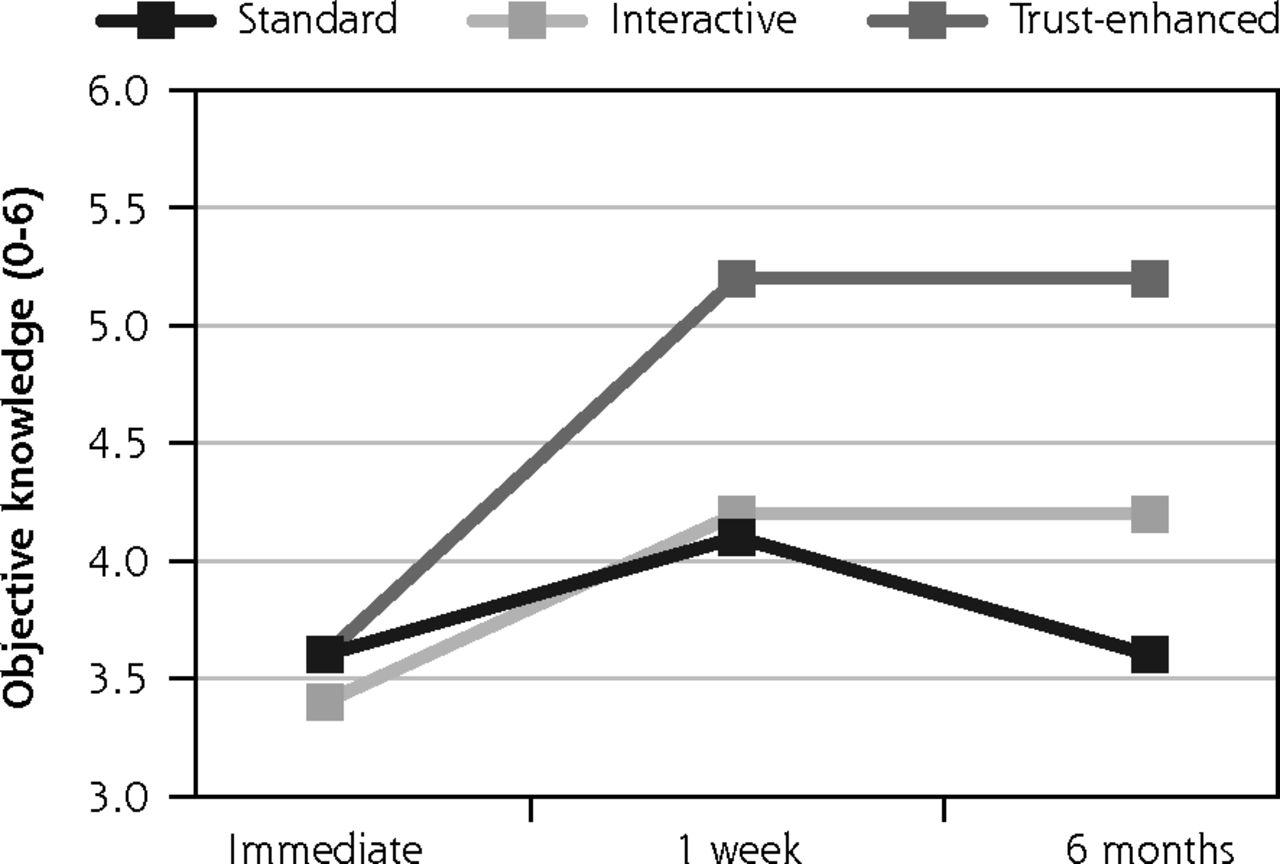
Secondary outcomes: objective knowledge, perceived voluntariness, and trust in medical researchers.
Notes: Values for all outcomes represent means by e-consent study arm by follow-up assessment time point based on descriptive analyses of study data. Numbers of participants for all outcomes were 734 at immediate follow-up, 624 at 1-week follow-up, and 510 at 6-month follow-up. Numbers of participants for at each follow-up by e-consent group are shown in Figure 1.
Likewise, mean subjective understanding of consent was similar between versions at immediate follow-up and at 1-week follow-up. However, at six month follow-up, mean understanding decreased to 57.6 (19.0) for the standard e-consent. There was less change for both the interactive version with a mean of 72.3 (13.4) and the trust-enhanced version with a mean of 89.6 (16.0).
Similar trends were observed for secondary outcomes, with 2 exceptions. For the trust-enhanced e-consent only, objective knowledge increased from 3.6 (1.3) at immediate follow-up to 5.2 (1.1) at the 1-week follow-up to and maintained this increase after 6 months. For the interactive e-consent, perceived voluntariness decreased slightly from the 1-week follow-up mean of 37.9 (5.5) to 36.7 (3.2) at the 6-month follow-up (Figure 3).
Perceived voluntariness was the only outcome for which the interactive e-consent was not superior to the standard version at 6-month follow-up.
Results of our multivariate regression analyses (Table 2) showed that increases from 1-week to 6-month follow-up in both satisfaction (B = 0.4, SE = 1.0; P <.001) and understanding (B = 18.0, SE = 2.6; P <.001) were significantly higher among participants for the interactive e-consent compared with the standard version. In other words, use of the interactive e-consent was associated with a 0.4-point higher increase in satisfaction (1 to 5) and an 18.0-point higher increase in understanding (0 to 100) compared with increases for users of the standard version after 6 months. Similar trends were observed when comparing the interactive e-consent with the standard version after 6 months on the secondary outcomes of objective knowledge (B = 1.2, SE = 0.2; P <.001) and trust in medical researchers (B = 6.6, SE = 0.9; P <.001). However, 6-month changes in perceived voluntariness did not differ significantly between the interactive and standard e-consents (B = 0.81, SE = 0.66; P = .22).
Adjusted Difference in Outcomes for Trust-Enhanced e-Consent Compared With the Standard and Interactive e-Consents at 6-Month Follow-Up
Finally, compared with the differences between the interactive and trust-enhanced e-consents at 1-week follow-up, the trust-enhanced version was further associated with significantly higher changes in satisfaction (B = 0.4, SE = 0.07; P <.001) and understanding (B = 14.09, SE = 2.42; P <.001). We also observed significantly higher increases for the trust-enhanced version from 1 week to 6 months for objective knowledge (B = 0.92, SE = 0.16; P <.001), perceived voluntariness (B = 3.18, SE = 0.68; P <.001), and trust in medical researchers (B = 5.9, SE = 0.90; P <.001), compared with increases for the interactive e-consent.
DISCUSSION
Six months after consenting to share their health record data for research, patients reported higher increases in satisfaction, subjective understanding, and other outcomes when they used an e-consent with interactive information or an e-consent with interactive information plus trust-enhancing messages, compared with patients who used a standard e-consent without these additional features. Together, our findings indicate that offering hyperlinked information on basic research concepts and additional facts about research protections may strengthen key aspects of informed consent. In particular, including these features in e-consents may ensure that potential research participants receive and understand information that the reasonable-person standard of disclosure requires. Importantly, we are not claiming that consent processes should be designed to increase recruitment or manipulate patients into participating in research. Instead, we argue that consent processes with these additional informational features can effectively address known sources of mistrust and poor comprehension and increase long-term satisfaction and understanding for more patients.
Notably, the effect of trust messaging on our study outcomes was manifest only at 6-month follow-up. Our previously reported analysis of immediate and 1-week outcomes showed no significant differences between e-consent versions.20 Also, while patients using the interactive e-consent reported improved outcomes on several measures, actual use of the interactive hyperlinks was rare (80 total clicks across the 491 participants who used the interactive or trust-enhanced e-consent). Thus, the observed effects cannot be attributed to actual receipt of the hyperlinked information. One potential mechanism is that the transparency inherent in the interactive and trust-enhanced e-consents had protective effects on the outcomes measured. For example, it is possible that participants across groups were exposed to news media involving data privacy breaches that occurred between the point of intervention and 6-month follow-up. In that case, use of the interactive or trust-enhanced e-consents may have been protective against attitudes of distrust or skepticism triggered by media coverage of these events. Regardless of whether they actually used the interactive links, we speculate that, compared with participants using the standard e-consent, participants who used the interactive or trust-enhanced e-consents may have had increased perceptions of researcher transparency at 6-month follow-up–simply because the information was made available to them in the e-consent. However, we are unable to further analyze this conjecture with the available data. Therefore, this phenomenon should be explored in more detail in future research.
Given that few previous studies have examined relevant outcomes of informed consent interventions beyond the short-term,21-24 our findings present novel insights into longer-term effects of consent processes on patients’ consent perceptions and experiences. Improved long-term satisfaction, understanding, trust, and other outcomes are particularly important in the context of broad consent, in which patients may have minimal or no contact with research staff after the initial point of consenting, and in which participants have consented to something that, in fact, persists over a long period of time. Given the unspecified nature of these future research studies, the language associated with broad consent may be frustratingly vague and general. Based on the findings of this trial, factual messages emphasizing privacy and other institutional research protections may be an effective way for researchers and institutions to elicit high levels of longer-term satisfaction, understanding, knowledge, voluntariness, and trust.
Significant practical and ethical challenges remain to widespread implementation of broad consent, including concerns about re-identification risks associated with patient data that has been certified as de-identified.25 However, given the difficulties associated with obtaining study-specific prospective consent, including the burden it places on patients, providers, and research staff, health information technology has the potential to facilitate effective and efficient administration of broad informed consent while honoring patient preferences for autonomy and transparency.13 Finally, given that our study evaluated the impact of interactive information and trust-enhancing features in the context of a broad consent mechanism, future research should examine the utility of these and other features in consenting patients for more complex or intensive research and clinical situations, such as procedures or interventional trials.
Limitations
This trial was conducted among English-speaking, family medicine patients in Florida who, on average, were middle-aged, female, non-Latinx White or Black adults. Results may not generalize to patients from other clinical settings, geographic areas, or demographic backgrounds. Related to this, our sample consisted of participants who agreed to participate in a research study about consent, who may be different in important ways from patients who declined to participate in this study. Furthermore, since the research consent process itself was under study, participant responses may not be fully representative of patients asked to consent to research uses of their health data in a real world scenario. In addition, it was not possible to blind data collectors to participant assignment. Finally, participants who were lost to follow-up may differ systematically from those who contributed data at all assessment points. However, we conducted a weighted dropout analysis and did not find significant differences in outcomes when comparing weighted and unweighted results. Additionally, overall rates of dropout did not differ between study arms. Still, we cannot rule out the possibility that unobserved differences between dropouts across the study arms biased our effect estimates.
Acknowledgments
The authors acknowledge the contributions of the University of Florida Department of Community Health and Family Medicine and the Department’s clinic staff for supporting the study. The authors also acknowledge the Regenstrief Institute software engineering team for its contributions to developing the e-consent application.
Footnotes
Conflicts of interest: Dr Harle has served as a paid consultant to the New York eHealth Collaborative (NYeC) and Indiana Health Information Exchange (IHIE). The remaining authors have no conflicts of interest to disclose.
To read or post commentaries in response to this article, see it online at https://www.AnnFamMed.org/content/19/1/16.
Funding support: This study was funded under grant number R01HD086700 by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD).
Previous presentations: American Medical Informatics Association (AMIA) 2019 Annual Symposium; November 16-20, 2019; Washington, DC.
Trial registration: “An Interactive Patient-Centered Consent for Research Using Medical Records”; NCT03063268; https://clinicaltrials.gov/ct2/show/NCT03063268
Supplemental materials: Available at https://www.AnnFamMed.org/content/19/1/16/suppl/DC1/.
- Received for publication January 20, 2020.
- Revision received April 27, 2020.
- Accepted for publication June 3, 2020.
- © 2021 Annals of Family Medicine, Inc.
References
In this issue



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.